This post is also available in:
![]() Español (Spanish)
Español (Spanish) ![]() Français (French)
Français (French)
Pronunciation: sɪstɪk faɪˈbroʊsɪs
What is cystic fibrosis?
Cystic fibrosis is a progressive genetic disease. It affects both males and females. Cystic fibrosis can cause a variety of problems that impact the health of a child’s lungs and a child’s ability to gain adequate nutrition.
Advancements in treatments have improved the quality and length of life for individuals with cystic fibrosis. In particular, nutrition has been a key factor in improving quality of life, lung function, growth, development, and survival. However, the disease may lead to premature death. The current average predicted survival age for someone with cystic fibrosis is 48 years.
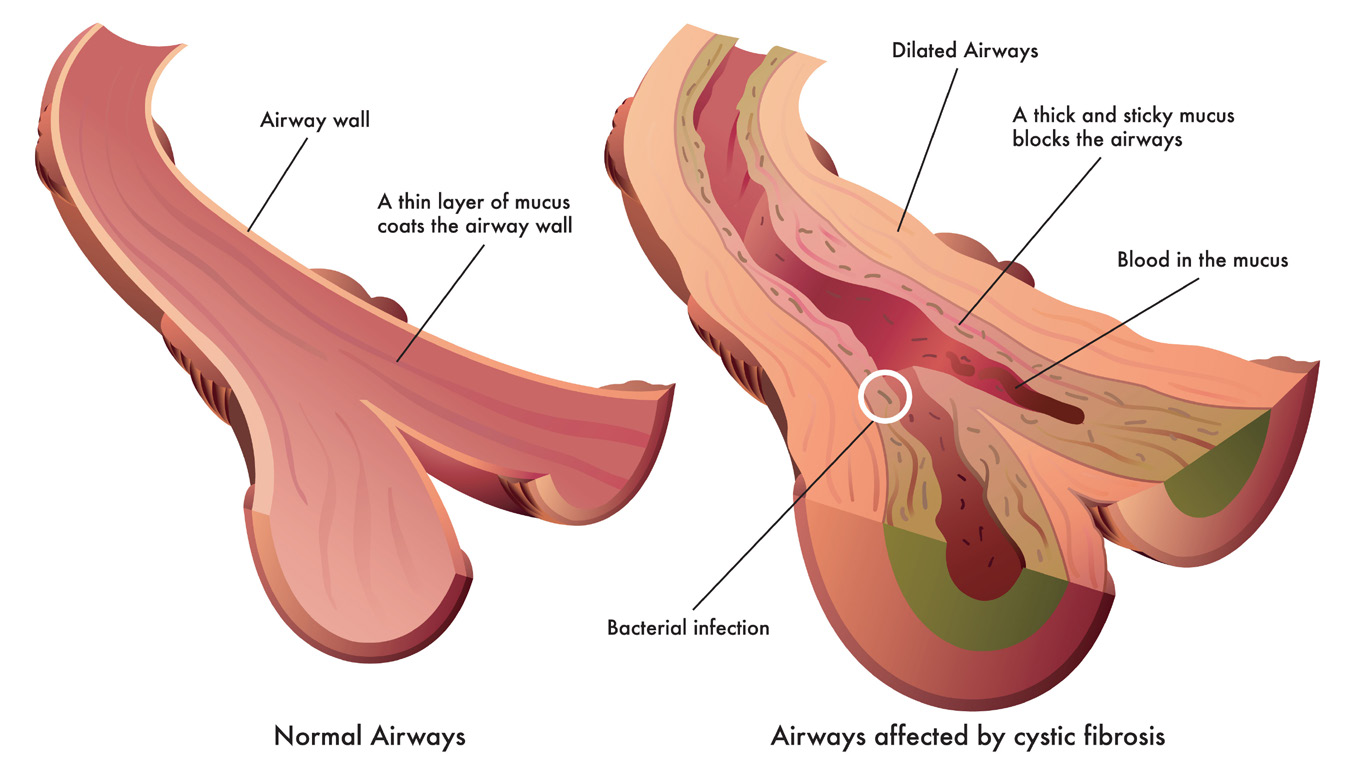
What causes cystic fibrosis?
Cystic fibrosis is caused by genetic mutations in a protein called CFTR (cystic fibrosis transmembrane conductance regulator). This protein is found in many cells of the body, but most importantly in cells of the respiratory and gastrointestinal tract.
CFTR controls how certain ions (chloride and bicarbonate ions) and fluids move across the cell’s membrane. Mutations in CFTR prevent the ions and fluids from being able to move correctly across the membrane. These problems can cause a thick mucus to build-up in the lungs, pancreas, intestines, and other organs. This mucus buildup causes infection and/or inflammation, damaging the organs and preventing them from working correctly.
How common is cystic fibrosis and who is at risk for having it?
The incidence of cystic fibrosis differs in groups of people with different ethnic backgrounds. Cystic fibrosis is most common in Caucasian Americans, occurring at a rate of 1:2,500 in the United States. It occurs less frequently in other ethnic groups: 1:13,500 in Hispanic Americans, 1:15,000 in African Americans, and 1:35,000 in Asian Americans.
What are the signs and symptoms of cystic fibrosis?
People with cystic fibrosis can have different symptoms, including salty-tasting skin, persistent coughing, frequent lung infections, soft growths (called polyps) in the lining of the nasal passages, poor weight gain, frequent abdominal pain, greasy or bulky stools, constipation, and/or inflammation in the pancreas (called pancreatitis).
In older individuals, cystic fibrosis can also cause recurrent pancreatitis. Both males and females with cystic fibrosis also might not be able to have children (called sterility).
How is cystic fibrosis diagnosed?
In the United States, screening for cystic fibrosis occurs shortly after birth through newborn screening. Only a few drops of blood are needed to test for multiple diseases, including cystic fibrosis.
In some states, this test is repeated a couple of weeks after birth. If the test is abnormal, a sweat chloride test is performed to confirm the diagnosis. Further genetic testing may be performed to determine if an individual has gene mutations.
Not all individuals are diagnosed at birth, with some being diagnosed as toddlers, teenagers, or even adults. If your healthcare provider thinks your child has symptoms associated with cystic fibrosis, they may order sweat chloride and genetic testing. You may also be referred to a healthcare center with cystic fibrosis specialists.
What tests are used in children to diagnose cystic fibrosis?
Most screening programs measure the level of immunoreactive trypsinogen (IRT) in the blood. This is normally found in small levels in the body, but individuals with cystic fibrosis usually have high levels of IRT.
Elevated IRT levels indicate that cystic fibrosis is possible. Genetic testing may then be performed. A sweat chloride test measures the amount of chloride ions in the sweat. This test is performed to make a final diagnosis of cystic fibrosis. Cystic Fibrosis Care Centers have expertise in performing and interpreting sweat chloride testing.
What is the treatment for cystic fibrosis?
Treatment plans for cystic fibrosis vary based on the symptoms and how severe the disease is in each patient. However, comprehensive cystic fibrosis treatment plans usually include visits to a Cystic Fibrosis Care Center every three months and daily oral or inhaled medications to keep the lungs healthy. Patients are often treated with antibiotics. Nutrition also is a very important part of therapy.
What can I expect if my child has cystic fibrosis?
Cystic fibrosis affects every individual differently, so the disease type and severity of symptoms can vary from one person to the next. You and your child will meet regularly (about every three months) with the Cystic Fibrosis Care Center team to keep your child as healthy as possible.
Children with cystic fibrosis often get more lung infections than unaffected children, so they need to do breathing treatments and take medicine to keep their lungs healthy.
Children with cystic fibrosis also usually have a harder time breaking down and absorbing nutrients from food, which can be treated by changing the diet and with medications, such as digestive enzymes. Nutrition supports lung function, growth, and both the quality and duration of life.
While there is no cure for cystic fibrosis, advancements in research and care are helping people with cystic fibrosis live long enough to realize their dreams of going to college, having a career, and having a family of their own.
Where can I find support for my child and my family?
Your healthcare providers can connect you with support in your local area. Every Cystic Fibrosis Care Center has a social worker that can help provide you with the resources you need. Cystic Fibrosis Care Centers have multiple providers across different specialties who all work together to care for their patients.
There are many organizations and online groups that support individuals and families with cystic fibrosis. Here are a few to get you started:
- Cystic Fibrosis Foundation: www.cff.org
- Cystic Fibrosis Research Inc: www.cfri.org
Can cystic fibrosis be prevented?
No. Because it is a genetic disease, there is nothing you can do to prevent cystic fibrosis.
Cystic fibrosis is an autosomal recessive disease, which means you must get one copy of the mutated cystic fibrosis gene from each parent. Often, parents do not know they have a copy of the mutated gene because they are not affected by the disease. These individuals are called “carriers.” When two carriers have a child, there is a 25% chance that the child will have cystic fibrosis.
The risk of being a carrier for cystic fibrosis varies by ethnicity, with Caucasian Americans at higher risk than African Americans, who are at greater risk than Asian Americans. Testing can be done to determine if parents are carriers.
Even though cystic fibrosis cannot be prevented, early diagnosis, consistent treatments, and regular visits with a cystic fibrosis medical specialist can help improve a patient’s quality of life and long-term prognosis.
What is the role of the gastroenterologist in caring for patients with cystic fibrosis?
While cystic fibrosis is thought of as a disease that primarily affects the lungs, most patients with cystic fibrosis also have digestive issues. This is because the gene mutations that cause cystic fibrosis also affect the ability of the pancreas (an important digestive organ) to digest food.
In addition, some patients with cystic fibrosis develop liver inflammation and are at risk for liver scarring, called cirrhosis. Therefore, most patients with cystic fibrosis are cared for by a multidisciplinary team that includes a pulmonologist (lung specialist) and a gastroenterologist (specialist in digestive issues, pancreas function, and liver disease).
Cystic fibrosis quick facts:
- Cystic fibrosis is a progressive genetic disease that affects children and adults.
- Cystic fibrosis is most common in Caucasians but also occurs in those of other ethnic backgrounds.
- Cystic fibrosis is diagnosed through a combination of blood, genetic, and sweat chloride tests.
- Treatment plans vary for each individual but usually require daily medicine and regular visits to a cystic fibrosis specialist every three months.
- Respiratory treatments, antibiotic use, and nutrition are very important parts of the therapy and can improve quality and duration of life.
- Advances in research and care are improving and extending the lives of individuals with cystic fibrosis, but many still experience shortened lifespans.
Author: Zachary M. Sellers, MD & Asim Maqbool, MD
Editor: Athos Bousvaros, MD
March 2021
This post is also available in:
![]() Español (Spanish)
Español (Spanish) ![]() Français (French)
Français (French)
