
This post is also available in:
![]() Español (Spanish)
Español (Spanish) ![]() Français (French)
Français (French)
What is a Polyp?
- A polyp is a small growth of extra tissue that forms from the lining of the intestine. Sometimes there may be no symptoms when the polyp is small. The polyps can grow large over time and lead to a blockage (obstruction) in the intestines which can cause abdominal pain or vomiting. Sometimes when they fall off, they can lead to blood in stool and low blood count (anemia).
- Polyps can form as a result of genetic mutations, which are changes to a person’s DNA, that can occur randomly or can be inherited and passed down through the family.
When should I bring my child to their provider if I am worried about a polyp?
It is always recommended to discuss any concerns you have with your child’s provider, such as if your child is having abdominal pain, vomiting, or blood in the stool. You will also want to let your provider know if there is a family history of polyps or colon cancer in a close relative (in either parent, siblings, grandparents, aunts, or uncles).
Familial Adenomatous Polyposis (FAP)
Diagnosis
What is FAP?
Familial adenomatous polyposis, known as “FAP” in short, is a polyposis syndrome that causes hundreds to thousands of pre-cancerous, adenomatous polyps in the gastrointestinal tract, especially the colon and rectum. Over time, these adenomas can transform into cancer, so monitoring with endoscopy is required.
What causes FAP?
In most cases, FAP is caused by a mutation in the adenomatous polyposis coli (APC) gene. Often, this mutation is inherited (passed down in the family). However, 3 out of 10 children with FAP are the first in their family to have FAP, and in some cases, no mutation is identified. Children with FAP usually develop numerous adenomatous polyps around adolescence – adenomatous polyps are a specific type of polyp that can transform into cancer over time. Children with adenomatous polyps are managed similarly regardless of whether a genetic mutation is identified.
Does the phenotype (disease presentation) depend on the genetic mutation?
Yes, there are mutations in the APC gene that cause more colonic polyps to form over time. Mutations are usually described by a number that corresponds to the position of the mutation (change in letter sequence) within the gene. These include mutations at certain locations, such as at codon 1309 and between codons 1250 and 1464. There is also a milder form of FAP called attenuated FAP, in which polyps can form later in life.
Are there any other associated conditions with FAP?
Aside from polyps in the colon, individuals with FAP are likely to develop polyps in other parts of the intestine. Outside of the intestine, children with FAP are at risk of developing a type of liver tumor (hepatoblastoma). They may have findings in the back of the eye - the retina called congenital hypertrophy retinal pigmentation epithelium (CHRPE) or pigmented ocular fundus lesions of FAP (POFL). Children may also have benign growths in bone and skin - osteomas and fibromas, and extra or missing teeth. People with FAP are also at higher risk of other cancers, desmoid tumors, thyroid cancer, and brain tumors.
Should first-degree relatives be screened for FAP?
Yes, genetic testing for the identified APC mutation should be offered to all first-degree relatives. If genetic testing has not been completed or the genetic mutation is unknown, then a colonoscopy is recommended annually, starting between 10 to 15 years of age. If no adenomas are found after 15 years of age, then a colonoscopy can be performed every two years or longer.
Management
When should the first upper endoscopy be performed for a child with FAP?
Upper endoscopy should occur around 20 to 25 years of age. Earlier evaluation should occur if there is a family history of duodenal cancer or concerning symptoms.
When should my child undergo their first colonoscopy?
For children with confirmed FAP and at-risk children in whom genetic testing is not possible, colonic surveillance should start between the ages of 12 to 14 years. Attenuated FAP is a milder form of the disease in which patients may not have as many adenomas and thus may not present until adulthood.
How often should surveillance be performed?
Surveillance colonoscopy every 1 to 3 years, depending on phenotype. Colonoscopy is indicated earlier if a child has blood in stools, anemia, or other symptoms of polyps.
What is the treatment of FAP?
Currently, there is no curative treatment for FAP. Children with FAP need to have close surveillance for polyps that develop pre-cancerous changes for removal with endoscopy. However, sometimes there are too many polyps to remove with endoscopy, and surgery may be required.
What are the types of surgeries performed for FAP?
There are three types of surgeries performed for children with FAP, all are intended to remove most or all of the colon:
- Total colectomy with ileorectal anastomosis (IRA)
- Proctocolectomy with ileal pouch anal anastomosis (IPAA)
- Proctocolectomy with end ileostomy
Due to the rarity of colon cancer in children, the timing of colectomy in children and adolescents under 18 years of age is not established. Even after colectomy, the remaining part of the rectum or pouch should be monitored every 6 to 12 months for the development of polyps and cancer.
What about other types of cancer?
There is a higher risk for thyroid cancer, and thyroid ultrasound should be obtained during the late teenage years and, if normal, then repeated every 2 to 5 years. There is also an association with hepatoblastoma, a type of liver cancer. Abdominal exams, ultrasound, and monitoring of AFP levels in the blood can occur every 3 to 6 months until 5 years of age.
There is no evidence for routine screening for brain tumors or pancreatic cancer, and this should be performed based on family history. While stomach and small intestinal cancer can occur, specialized surveillance is not recommended unless there are advanced polyps in the stomach or small intestine. For patients with FAP and a family history of desmoid tumors, an abdominal MRI every 3 to 5 years after colectomy should be done to screen for abdominal desmoid tumors.
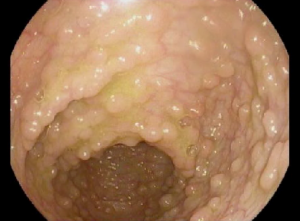
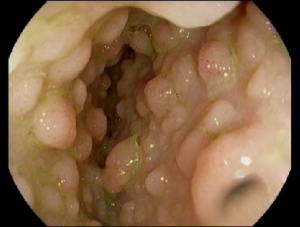
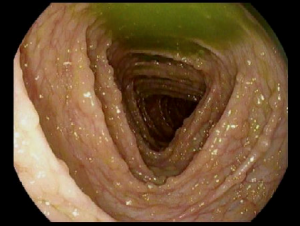
Peutz-Jeghers Syndrome (PJS)
Diagnosis
What is PJS?
Peutz-Jeghers syndrome (PJS) is a rare inherited (passed down from a family member) condition associated with polyps, dark-colored spots, and an increased risk of certain types of cancer. It occurs in approximately 1 in 200,000 people.
Children with PJS can have polyps form in their gastrointestinal (GI) tract (stomach, small intestine, and large intestine) as well as in the nose, lungs, and bladder. These polyps are considered hamartomatous polyps because they possess specific features when viewed under a microscope.
Hamartomatous polyps are benign (not cancerous) overgrowths of tissue. PJS is also associated with characteristic freckling or dark spots (muco-cutaneous pigmentation) around the lips and other parts of the body (mouth, nostrils, hands, feet, perianal area) that may fade over time.
The diagnosis is made in a person with:
- Two or more PJS polyps on endoscopy
- Any number of PJS polyps with a family history of PJS
- Characteristic freckling (on the lips and in the finger-nail bed) with a family history of PJS
- Any number of PJS polyps and characteristic freckling
What causes PJS?
PJS is caused by a mutation (change) in the STK11 gene that is responsible for controlling cell growth. PJS is caused when there is a mutation in one copy of this gene; however, not every patient with PJS has a mutation in the STK11 gene. Approximately 1 in 2 of children with PJS have a parent with the syndrome, and the other half the time, children may be the first one in their family affected.
Does the phenotype (disease presentation) depend on the genetic mutation?
There is no clear relationship between the gene mutation (genotype) and the disease presentation (phenotype). However, there is variability among individuals in the timing and types of disease presentation (phenotype) that can be caused by the development of polyps. These disease presentations may include anemia due to gastrointestinal bleeding from the polyps, abdominal pain, small bowel obstruction (blockage) or intussusception (where the polyp can cause telescoping of the bowel within itself), leading to pain, vomiting, and sometimes a full blockage.
Are there any other associated conditions with PJS?
Children and adults with PJS are at risk of other disease manifestations outside the intestine – in children, these include testicular and ovarian tumors, for which regular ultrasound testing is needed. In adults, there is also the risk of cervical, breast, ovarian, endometrial cancer, and pancreatic cancer.
Should first-degree relatives be screened for PJS?
Yes, genetic testing should be offered to all first-degree relatives. The vast majority of patients with PJS have the STK11 mutation detected, but if there has not been a gene detected in a family member, then genetic testing in children cannot be performed.
Management
When should an endoscopy be performed for a child with PJS?
Investigation of the GI tract is recommended to start no later than eight years of age unless symptoms arise earlier. The investigations would include upper endoscopy (which looks at the esophagus, stomach, and first part of the small intestine) and colonoscopy (which looks at the large intestine to the last part of the small intestine). Since the scope cannot view the entire small intestine, other methods are used to view the remainder of the small intestine, including MRI (magnetic resonance imaging) or video capsule endoscopy (a pill with a camera that moves through the GI tract).
How often should surveillance be performed?
Surveillance of the GI tract needs to be individualized depending on symptoms, risks of complications from the polyps versus risks from the testing, and family perspective. It is very important to have discussions with your genetic counselor and GI doctor about what symptoms to look out for (bleeding, abdominal pain, vomiting) so that children can have sooner evaluation if needed.
What is the treatment of PJS?
Currently, there is no preventative or curative treatment for PJS. Children with PJS need to have close surveillance for polyps that may cause complications like bleeding, intussusception, or blockage. If possible, the goal is to remove large polyps to prevent these complications, which may be possible with endoscopy. However, sometimes the complications may occur unexpectedly and require immediate surgery.
Patients with a blockage or intussusception require urgent surgery. At the time of surgery, it is possible to assess the remainder of the small intestine using a scope during the surgery to try to identify and remove additional large polyps.
What about cancer risk in PJS?
Cancer during childhood in PJS is very rare. Unfortunately, there is a lifetime increased risk of cancer beginning in adulthood. In children, it is important to have regular monitoring and physical examinations for signs of early puberty, which may indicate a rare tumor of the ovary (in girls) or testicles (in boys). For boys, annual testicular exams and observation for any signs of early puberty or increased breast size should begin at diagnosis.
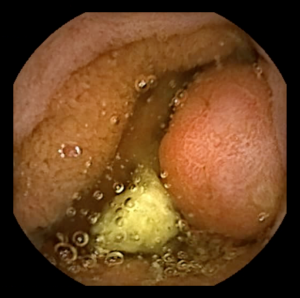
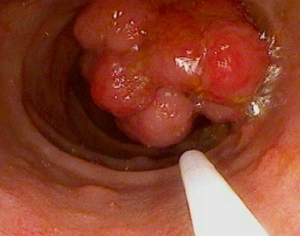
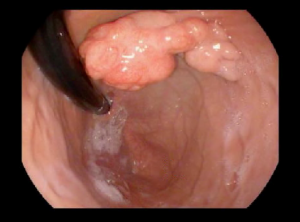
Juvenile Polyposis Syndrome
Diagnosis
What is JPS?
Juvenile Polyposis Syndrome (or JPS) is a rare condition that results in the formation of juvenile (hamartomatous) polyps in the gastrointestinal tract and is associated with an increased risk of colorectal cancer. It affects approximately 1 in 130,000 people.
This syndrome is different from the solitary juvenile polyps that are the most common type of polyp in children and do not have an associated cancer risk.
The diagnosis of JPS is made when five or more juvenile polyps are found in the colon or rectum, or in a person with a family history of JPS and any number of juvenile polyps, or any juvenile polyp found in other parts of the gastrointestinal tract.
What causes JPS?
JPS is caused by a defect (mutation) in gene function. There are two mutations implicated, SMAD4 on chromosome 18 and BMPR1A on chromosome 10. It is inherited in an autosomal dominant pattern, meaning it passes from generation to generation. However, only approximately 1 in 2 children have a defect in their genes. The other 50% meet the criteria for diagnosis, but there is not a genetic mutation identified on testing.
Patients without an identified gene mutation have none or few polyps in the stomach and small intestine, and the number of polyps in the colon decreases after the first decade of life. These patients also tend to present earlier in life, have a negative family history, and have a lower risk of colon resection.
Does the phenotype (disease presentation) depend on the genetic mutation?
Yes, although this is still unclear, patients with the SMAD4 mutation have more polyps in the stomach and a higher risk of cancer. Also, some patients with this genetic mutation may have what is called hereditary hemorrhagic telangiectasia (HHT). These patients have abnormal blood vessels in various organs, including the brain, lungs, and liver, and usually present with nosebleeds that are difficult to control.
There is a severe form of JPS called Juvenile Polyposis of Infancy of JPI. These patients also have another mutation on the same chromosome 10 called PTEN. It presents in the first two years of life and has a more aggressive presentation with diarrhea, low protein, intestinal bleeding, poor weight gain, and usually requires removal of the colon at an earlier age.
Are there any other associated conditions?
There are no known allergic or autoimmune conditions associated with JPS as of now.
Should first-degree relatives be screened for JPS?
Yes, when a genetic mutation has been identified, family members should be tested. If no mutation is identified, it is recommended that parents and siblings 15 years old and older undergo colonoscopy to evaluate for juvenile polyps.
Management
When should an endoscopy be performed for a child with JPS?
Lower endoscopy (colonoscopy) is recommended to start at 12-15 years of age. Upper endoscopy is not required before 18 years of age unless symptoms arise earlier.
How often should surveillance be performed?
Surveillance is necessary to prevent bleeding, anemia, abdominal pain, and cancer prevention. It is recommended to have a colonoscopy yearly if polyps are found and until all large polyps have been removed, after that, every 1 to 5 years.
What is the treatment of JPS?
There is no specific treatment for JPS besides the removal of the polyps, also called polypectomy.
In patients with the severe form (JPI), a medication called sirolimus has been shown to improve anemia and low albumin and reduce the risk of colon removal.
What about cancer risk in JPS?
Overall, the risk of malignant transformation in children with JPS is rare, but it has a lifetime increased risk of 10 to 50% of colon or rectal cancer. Therefore, surveillance with colonoscopy and removal of the large polyps is important.
Children with the severe form (JPI) have an increased risk of thyroid cancer, breast cancer, endometrial cancer, and other cancer types later in life due to PTEN gene mutation, and surveillance is recommended to start with thyroid ultrasound at 7 years old.
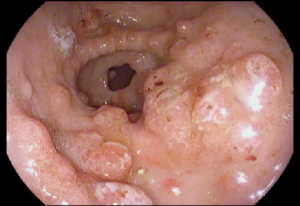
General & Miscellaneous Questions
Is there a standard regimen for bowel clean-out prior to colonoscopy?
A bowel preparation is required prior to a colonoscopy. There is no standard protocol universally followed in pediatrics. Each pediatric gastroenterology program creates its own protocol, which may be different from another practice.
How often should surveillance for small intestinal polyps be performed?
The type of test and frequency for testing for small intestinal polyps depends on the syndrome and characteristics related to the patient. In general, patients with Peutz-Jeghers Syndrome need small bowel surveillance (periodic checking for polyps) because these polyps can cause obstruction. Your gastroenterologist will guide you to either get a capsule endoscopy in your child or a special MRI of the abdomen. The frequency varies between 1 and 3 years usually and is determined, in part, by how many polyps were noted during earlier procedures.
Which type of surgery, partial or total colectomy, is recommended for patients with polyposis?
Patients with polyposis require colectomy to prevent the development of cancer (in FAP) or, in some cases, to control the number of polyps that could not be removed endoscopically (Juvenile Polyposis or Peutz-Jeghers Syndrome). In patients with FAP, either a total colectomy with a pouch is planned (IPAA) or removal of the colon less the rectum to which the small bowel is attached – ileoanal anastomosis(IRA). The decision on which type of colectomy, or other types of colectomies, has to be made keeping in mind many factors, including the family history, age and gender of the child, and the type of gene mutation if known.
Is there a role for fecal calprotectin in the prediction or monitoring of polyp progression?
Fecal calprotectin is a protein that can be detected on a stool test, it is released by certain immune system cells in the intestine and can be a marker of inflammation in the intestines. Calprotectin may be helpful in prediction of polyp presence, especially for juvenile polyps, but does not differentiate from other causes of abdominal pain or bloody stools like infection or inflammatory bowel disease (IBD). Further studies are needed to determine whether calprotectin can be used to predict development or progression of polyps.
Is there a role for alternative or complementary therapies for polyps?
There are very limited studies evaluating complementary therapies in polyposis. Further study is needed to determine if these are safe and effective for use in children.
What role does the gut microbiome play in polyp formation and progression?
There is limited evidence of the role of gut microbiome in progression of colonic polyps and development of colorectal cancer. There are currently no diagnostic or therapeutic implications of gut microbiome in polyposis disorders.
Is there a role for dietary therapy for polyps?
There have been many studies using animal models of FAP as well as in human patients with adenomas at an older age but none, thus far in children with FAP. A healthy balanced diet including fiber is important but the beneficial impact of fish oil and probiotics, although likely, remains unproven in this population. The impact of other, specific supplements such as turmeric or resveratrol is also unclear and is not routinely recommended.
Resources
How do I find a pediatric gastroenterologist with an interest or expertise in polyposis?
Optimal care for patients with polyposis syndromes includes a multidisciplinary team comprised of a pediatric gastroenterologist, pediatric oncologist, pediatric surgeon, and genetics counselor.
What are the opportunities to participate in research?
Please ask your pediatric gastroenterologist, oncologist and genetics counselor about additional opportunities to participate in research at your center.
Is the COVID vaccine safe for children with polyps?
Yes, the COVID vaccination is not a contraindication and can be safely administered.
Author: Claudia C. Phen, MD ; Thomas Attard, MD; Sabina Mir, MD; Isabel Rojas, MD; Mary Zachos, MD
Editor: Christine Waasdorp Hurtado, MD, MSCS, FAAP
March 2023
This post is also available in:
![]() Español (Spanish)
Español (Spanish) ![]() Français (French)
Français (French)

