
This post is also available in:
![]() English (Anglais)
English (Anglais) ![]() Español (Espagnol)
Español (Espagnol)
Qu’est-ce qu’un polype ?
- Un polype est une petite excroissance de tissu qui se forme dans la paroi de l'intestin. Parfois, il n'y a pas de symptômes lorsque le polype est petit. Le polype peut croître au fil du temps et causer une obstruction dans l’intestin, ce qui peut causer des douleurs abdominales ou des vomissements. Un polype qui se détache risque de causer du sang dans les selles et une anémie.
- Un polype peut résulter d’une mutation génétique, à savoir, une modification de l’ADN d’une personne qui peut survenir de manière aléatoire ou être héritée et transmise dans la famille.
Quand dois-je faire examiner mon enfant par son médecin si je m'inquiète de la présence d'un polype ?
- Il est toujours conseillé de vous adresser au médecin de votre enfant ; par exemple, en cas de douleurs abdominales, de vomissements ou de présence de sang dans les selles. Faites aussi savoir au médecin s'il y a des antécédents familiaux de polypes ou de cancer du côlon chez un parent proche (l'un des parents, un frère ou une sœur, un grand-parent, une tante ou un oncle).
La polypose adénomateuse familiale
Diagnostic
Qu’est-ce la polypose adénomateuse familiale ?
La polypose adénomateuse familiale est un syndrome de polypose qui provoque des centaines, voire des milliers de polypes adénomateux précancéreux dans le tractus gastro-intestinal, en particulier dans le côlon et le rectum. Avec le temps, ces adénomes peuvent se transformer en cancer, d'où la nécessité d'une surveillance par endoscopie.
Qu’est-ce-qui cause la polypose adénomateuse familiale ?
Dans la plupart des cas, la polypose adénomateuse familiale est causée par une mutation du gène de la polypose adénomateuse du côlon. Souvent, cette mutation est transmise dans la famille. Cependant, 3 enfants sur 10 atteints de la polypose adénomateuse familiale sont les premiers de la famille à être atteints de la maladie, et dans certains cas, aucune mutation n'est identifiée. Un enfant atteint de la polypose adénomateuse familiale développe généralement de nombreux polypes adénomateux dans l'adolescence, un type particulier de polypes qui peut se transformer en cancer avec le temps. Un enfant atteint de polypes adénomateux est pris en charge de la même manière, qu'une mutation génétique ait été identifiée ou non.
Le phénotype (présentation de la maladie) dépend-il de la mutation génétique ?
Oui, il existe des mutations du gène de la polypose adénomateuse colique qui entraînent la formation d'un plus grand nombre de polypes du côlon au fil du temps. Les mutations sont généralement indiquées par un chiffre qui correspond à la position de la mutation (changement de la séquence de lettres) dans le gène. Il s'agit notamment de mutations à certains endroits, tels que le codon 1309 et entre les codons 1250 et 1464. Il existe également une forme plus faible de Polypose adénomateuse familiale, appelée Polypose adénomateuse familiale atténuée, dans laquelle les polypes peuvent se former plus tard dans la vie.
Existe-t-il d'autres pathologies associées à la Polypose adénomateuse familiale ?
Outre les polypes du côlon, les personnes atteintes de Polypose adénomateuse familiale sont susceptibles de développer des polypes dans d'autres parties de l'intestin. Un enfant atteint de Polypose adénomateuse familiale risque de développer un type de tumeur du foie (hépatoblastome). Il peut avoir une anomalie au fond de l'œil - la rétine - appelée hypertrophie congénitale de l'épithélium pigmentaire rétinien ou des lésions pigmentées de Polypose adénomateuse familiale dans le fond de l'œil. L’enfant peut également présenter des excroissances osseuses et cutanées bénignes (ostéomes et fibromes), ainsi que des dents supplémentaires ou manquantes. Un individu atteint de Polypose adénomateuse familiale est également plus exposé à d'autres cancers, aux tumeurs desmoïdes, au cancer de la thyroïde et aux tumeurs cérébrales.
Les parents au premier degré doivent-ils faire l'objet d'un dépistage de la Polypose adénomateuse familiale ?
Oui, un test génétique pour la mutation de la polypose adénomateuse du côlon identifiée doit être proposé à tous les membres proches de la famille. Si le test génétique n'a pas été effectué ou si la mutation génétique est inconnue, une coloscopie est recommandée chaque année à partir de l'âge de 10 à 15 ans. Si aucun adénome n'est détecté après l'âge de 15 ans, la coloscopie peut être effectuée tous les 2 ans ou au-delà.
Gestion
Quand la première endoscopie supérieure doit-elle être effectuée chez un enfant atteint de polypose adénomateuse familiale ?
L'endoscopie supérieure doit être pratiquée vers l'âge de 20 à 25 ans. Une évaluation doit être effectuée plus tôt en cas d'antécédents familiaux de cancer du duodénum ou de symptômes inquiétants.
Quand mon enfant doit-il subir sa première coloscopie ?
Pour l’enfant dont la polypose adénomateuse familiale est confirmée et l’enfant à risque pour lesquels un test génétique n'est pas possible, la surveillance du côlon doit commencer entre 12 et 14 ans. La polypose adénomateuse familiale atténuée est une forme plus faible de la maladie dans laquelle le patient n'a pas autant d'adénomes et qui ne se manifeste donc pas avant l'âge adulte.
À quelle fréquence faut-il effectuer une surveillance ?
Une coloscopie de surveillance est requise tous les 1 à 3 ans en fonction du phénotype. La coloscopie est indiquée plus tôt si l'enfant a du sang dans les selles, une anémie ou d'autres symptômes de polypes.
Comment traite-t-on la polypose adénomateuse familiale ?
À l'heure actuelle, il n'existe pas de traitement curatif pour la polypose adénomateuse familiale. L’enfant atteint de polypose adénomateuse familiale doit faire l'objet d'une surveillance étroite pour détecter les polypes qui développent des modifications précancéreuses et qui doivent être enlevés par endoscopie. Cependant, il arrive que l'endoscopie ne permette pas d'enlever suffisamment de polypes et qu'une intervention chirurgicale devienne nécessaire.
Quels sont les types d'interventions chirurgicales pratiquées en cas de polypose adénomateuse familiale ?
Il existe trois types d'interventions chirurgicales pour l’enfant atteint de polypose adénomateuse familiale, toutes visant à retirer la plus grande partie ou la totalité du côlon :
- une colectomie totale avec anastomose iléorectale ;
- une proctocolectomie avec anastomose anale par poche iléale ;
- une proctocolectomie avec iléostomie terminale.
En raison de la rareté du cancer du côlon chez les enfants, le timing de la colectomie chez les enfants et les adolescents de moins de 18 ans n'est pas établi. Même après une colectomie, la partie non-réséquée du rectum ou de la poche doit être surveillée tous les 6 à 12 mois pour détecter l'apparition de polypes et de cancers.
Qu’en est-il d’autres types de cancer ?
Le risque de cancer de la thyroïde est plus élevé et une échographie de la thyroïde devrait être réalisée à la fin de l'adolescence et, si normale, répétée tous les 2 à 5 ans. Il existe également un lien avec l'hépatoblastome, un type de cancer du foie. Un examen abdominal, une échographie et un contrôle des taux de polypose adénomateuse familiale dans le sang peuvent être effectués tous les 3 à 6 mois jusqu'à l'âge de 5 ans.
Il n'existe pas de données probantes sur le dépistage systématique des tumeurs cérébrales ou du cancer du pancréas, qui devrait être effectué en fonction des antécédents familiaux. Bien que le cancer de l'estomac et de l'intestin grêle puisse se produire, une surveillance particulière n'est pas recommandée, sauf en cas de polypes à un stage avancé dans l'estomac ou l'intestin grêle. Chez les patients souffrant de polypose adénomateuse familiale et ayant des antécédents familiaux de tumeurs desmoïdes, une IRM abdominale doit être réalisée tous les 3 à 5 ans après la colectomie pour dépister les tumeurs desmoïdes abdominales.
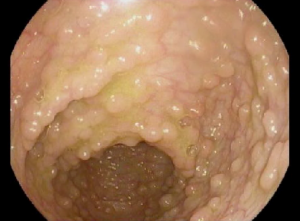
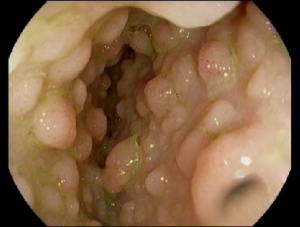
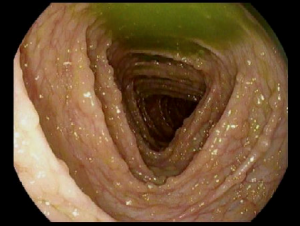
Syndrome de Peutz-Jeghers
Diagnostic
Qu’est-ce le Syndrome de Peutz-Jeghers ?
Le syndrome de Peutz-Jeghers est une maladie héréditaire rare (transmise par un membre de la famille) associée à des polypes, des taches de couleur foncée et un risque accru de certains types de cancer. Elle touche environ 1 personne sur 200 000.
Un enfant atteint du syndrome de Peutz-Jeghers peut avoir des polypes qui se forment dans son tractus gastro-intestinal (estomac, intestin grêle et gros intestin) ainsi que dans le nez, les poumons et la vessie. Ces polypes sont considérés comme des polypes hamartomateux car ils présentent des caractéristiques spécifiques lorsqu'ils sont observés au microscope.
Les polypes hamartomateux sont des excroissances de tissu bénignes (non cancéreuses). Le syndrome de Peutz-Jeghers est également associé à des taches de rousseur ou à des taches sombres (pigmentation muco-cutanée) caractéristiques autour des lèvres et d'autres parties du corps (bouche, narines, mains, pieds, zone périanale) qui peuvent s'estomper avec le temps.
Le diagnostic est établi chez une personne présentant :
- 2 polypes du syndrome de Peutz-Jeghers ou plus à l'endoscopie ;
- un nombre indéterminé de polypes du syndrome de Peutz-Jeghers avec des antécédents familiaux du syndrome ;
- des taches caractéristiques (sur les lèvres et dans le lit des ongles) avec des antécédents familiaux du syndrome ;
- un nombre indéterminé de polypes du syndrome de Peutz-Jeghers et des taches caractéristiques.
Qu’est-ce qui cause le syndrome de Peutz-Jeghers ?
Le syndrome de Peutz-Jeghers est dû à une mutation (changement) du gène STK11, responsable du contrôle de la croissance cellulaire. Le syndrome est causé par une mutation d'une copie de ce gène ; cependant, tous les patients atteints du syndrome ne présentent pas de mutation du gène STK11. Environ un enfant sur deux atteint du syndrome de Peutz-Jeghers a un parent atteint de ce syndrome et, dans l'autre moitié des cas, l'enfant peut être le premier de sa famille à être touché.
Le phénotype (présentation de la maladie) dépend-il de la mutation génétique ?
Il n'y a pas de relation claire entre la mutation du gène (génotype) et la présentation de la maladie (phénotype). Cependant, il existe une variation entre les individus en ce qui concerne le timing et la présentation de la maladie (phénotype) qui peuvent être causés par le développement de polypes. La maladie peut se manifester par une anémie due à des saignements gastro-intestinaux provoqués par les polypes, des douleurs abdominales, une obstruction de l'intestin grêle (blocage) ou une intussusception (lorsque le polype peut provoquer un télescopage de l'intestin), entraînant des douleurs, des vomissements et parfois une obstruction complète.
Existe-t-il d'autres pathologies associées au syndrome de Peutz-Jeghers ?
Les enfants et les adultes atteints du syndrome de Peutz-Jeghers risquent de présenter d'autres manifestations de la maladie en dehors de l'intestin - chez les enfants, il s'agit notamment de tumeurs testiculaires et ovariennes qui requièrent des examens échographiques réguliers. Chez les adultes, il y a également un risque de cancer du col de l'utérus, du sein, des ovaires, de l'endomètre et du pancréas.
Les membres de la famille au premier degré doivent-ils faire l'objet d'un dépistage du syndrome de Peutz-Jeghers ?
Oui, un test génétique doit être proposé à chaque membre de la famille au premier degré. La mutation STK11 est détectée dans la vaste majorité des patients atteints du syndrome, mais si aucun gène n'a été détecté chez un membre de la famille, il n'est pas possible de procéder à des tests génétiques chez un enfant.
Gestion
Quand doit on effectuer une endoscopie chez un enfant souffrant du syndrome de Peutz-Jeghers ?
On recommande de commencer l'examen du tractus gastro-intestinal au plus tard à l'âge de 8 ans, à moins que les symptômes ne se manifestent plus tôt. Les examens comprennent une endoscopie supérieure (qui examine l'œsophage, l'estomac et la première partie de l'intestin grêle) et une coloscopie (qui examine le gros intestin jusqu'à la dernière partie de l'intestin grêle). Comme l'endoscope ne permet pas de visualiser l'ensemble de l'intestin grêle, d'autres méthodes sont utilisées pour visualiser le reste de l'intestin grêle, notamment l'IRM (imagerie par résonance magnétique) ou l'endoscopie par capsule vidéo (capsule munie d'une caméra qui se déplace dans le tractus gastro-intestinal).
À quelle fréquence doit-on effectuer la surveillance ?
La surveillance du tractus gastro-intestinal doit être individualisée en fonction des symptômes, des risques de complications liés aux polypes par rapport aux risques liés au dépistage, et de la perspective familiale. I Il est très important de discuter avec votre conseiller génétique et votre médecin gastro-entérologue des symptômes à surveiller (saignements, douleurs abdominales, vomissements) afin que l'enfant puisse bénéficier d'une évaluation plus rapide si nécessaire.
Comment traite-t-on le syndrome de Peutz-Jeghers ?
Il n'existe à ce jour aucun traitement préventif ou curatif du syndrome. Un enfant atteint du syndrome doit faire l'objet d'une surveillance étroite afin de déceler la présence de polypes susceptibles de provoquer des complications telles que des hémorragies, des intussusceptions ou des obstructions. Si possible, l'objectif est d'enlever les gros polypes afin de prévenir ces complications, ce qui est faisable par endoscopie. Cependant, des complications peuvent parfois survenir de manière inattendue et nécessiter une intervention chirurgicale immédiate.
Le patient qui souffre d'une occlusion ou d'une intussusception doit être opéré d'urgence. Au moment de l'opération, il est possible d'évaluer le reste de l'intestin grêle à l'aide d'un scope pendant l'opération pour essayer d'identifier et d'enlever d'autres gros polypes.
Quels sont les risques de cancer liés au syndrome de Peutz-Jeghers ?
Il est très rare que le syndrome cause un cancer pendant l'enfance. Malheureusement, il existe un risque accru de cancer à l'âge adulte. Chez les enfants, il est important d'effectuer un suivi régulier et des examens physiques pour détecter les signes de puberté précoce qui peuvent indiquer une tumeur rare des ovaires (chez les filles) ou des testicules (chez les garçons). Chez les garçons, l'examen annuel des testicules et l'observation de tout signe de puberté précoce ou d'augmentation de la taille des seins doivent commencer dès le diagnostic.
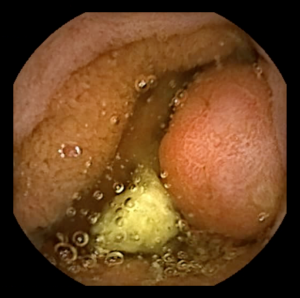
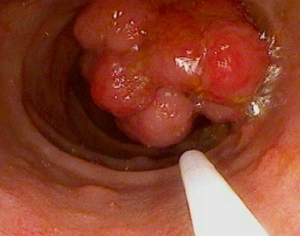
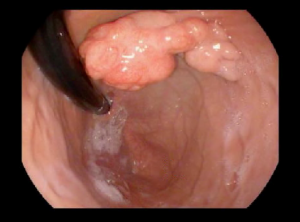
Syndrome de la Polypose juvénile
Diagnostic
Qu’est-ce le syndrome de la Polypose juvénile ?
Le syndrome de la Polypose juvénile est une maladie rare qui entraîne la formation de polypes juvéniles (hamartomateux) dans le tractus gastro-intestinal et qui est associée à un risque accru de cancer colorectal. Elle touche environ 1 personne sur 130 000.
Ce syndrome est différent des polypes juvéniles solitaires qui sont le type de polypes le plus courant chez les enfants et qui ne sont pas associés à un risque de cancer.
Le diagnostic du syndrome de la Polypose juvénile est posé lorsqu’au moins 5 polypes juvéniles sont découverts dans le côlon ou le rectum, ou chez une personne ayant des antécédents familiaux du syndrome et un nombre indéterminé de polypes juvéniles, ou tout polype juvénile trouvé dans d'autres parties du tractus gastro-intestinal.
Qu’est-ce-qui cause le syndrome de la Polypose juvénile ?
Le syndrome de la Polypose juvénile est causé par un défaut (une mutation) dans le fonctionnement d'un gène. Deux mutations sont impliquées, SMAD4 sur le chromosome 18 et BMPR1A sur le chromosome 10. Il est hérité selon un modèle autosomique dominant, ce qui signifie qu'il se transmet de génération en génération. Cependant, seul environ un enfant sur deux présente une anomalie dans ses gènes. Les autres enfants répondent aux critères de diagnostic, mais les tests ne permettent pas d'identifier une mutation génétique.
Le patient qui ne présente pas de mutation génétique identifiée n'a pas ou peu de polypes dans l'estomac et l'intestin grêle, et le nombre de polypes dans le côlon diminue après la première décennie. Un tel patient a également tendance à manifester le syndrome plus tôt dans la vie, à avoir des antécédents familiaux négatifs et à présenter un risque plus faible de résection du côlon.
Le phénotype (présentation de la maladie) dépend-il de la mutation génétique ?
Oui, le patient présentant la mutation SMAD4 a plus de polypes dans l'estomac et un risque plus élevé de cancer, bien que cela ne soit pas encore déterminé. Par ailleurs, certains patients porteurs de cette mutation génétique peuvent être atteints de ce que l'on appelle la télangiectasie hémorragique héréditaire. Ces patients ont des vaisseaux sanguins anormaux dans divers organes, notamment le cerveau, les poumons et le foie, et ont généralement des saignements de nez difficiles à contrôler.
Il existe une forme grave du syndrome de la Polypose juvénile appelée Polypose juvénile de l'enfance. Ces patients ont également une autre mutation sur le même chromosome 10 appelée PTEN. La maladie se manifeste au cours des deux premières années de la vie et est plus agressive, avec des diarrhées, une faible teneur en protéines, des saignements intestinaux et une faible prise de poids. Elle nécessite généralement l'ablation du côlon à un âge plus précoce.
Existe-t-il d’autres pathologies associées ?
À ce jour, il n'existe aucune pathologie allergique ou auto-immune connue associée au syndrome de la Polypose juvénile.
Les parents au premier degré doivent-ils faire l'objet d'un dépistage du syndrome de la Polypose juvénile ?
Oui, les membres de la famille doivent être testés lorsqu'une mutation génétique a été identifiée. Si aucune mutation n'est identifiée, on recommande que les parents et les frères et sœurs âgés de plus de 15 ans subissent une coloscopie afin d'évaluer la présence de polypes juvéniles.
Gestion
Quand un enfant atteint du syndrome de la Polypose juvénile doit-il subir une endoscopie ?
On recommande une endoscopie inférieure (coloscopie) à partir de l'âge de 12-15 ans. L'endoscopie supérieure n'est pas nécessaire avant l'âge de 18 ans, sauf si les symptômes apparaissent plus tôt.
À quelle fréquence la surveillance doit-elle être effectuée ?
La surveillance est nécessaire pour prévenir les hémorragies, l'anémie, les douleurs abdominales et le cancer. Il est recommandé de subir une coloscopie tous les ans si des polypes sont découverts et jusqu'à ce que tous les gros polypes aient été enlevés, puis tous les 1 à 5 ans.
Comment traite-t-on le syndrome de la Polypose juvénile ?
Il n'existe pas de traitement spécifique pour le syndrome de la Polypose juvénile, hormis l'ablation des polypes, également appelée polypectomie.
Chez le patient atteint de la forme grave de la maladie (Polypose juvénile de l'enfance), un médicament appelé sirolimus a démontré une amélioration de l’anémie et du faible taux d’albumine, et une réduction du risque d'ablation du côlon.
Qu'en est-il du risque de cancer lié au syndrome de la Polypose juvénile ?
En général, le risque de transformation maligne chez l’enfant atteint du syndrome est rare, mais le risque de cancer du côlon ou du rectum augmente de 10 à 50 % au cours de la vie. De ce fait, il est important d'effectuer une surveillance par coloscopie et d'enlever les gros polypes. Un enfant atteint de la forme grave (Polypose juvénile de l’enfance) présente un risque accru de cancer de la thyroïde, du sein, de l'endomètre et d'autres types de cancer plus tard dans la vie en raison de la mutation du gène PTEN. Il est donc recommandé de commencer la surveillance par une échographie de la thyroïde à l'âge de 7 ans.
Questions diverses
Existe-t-il un processus standard de nettoyage des intestins avant une coloscopie ?
Une préparation intestinale est nécessaire avant une coloscopie. Il n'existe pas de protocole standard universellement suivi en pédiatrie. Chaque programme de gastroentérologie pédiatrique crée son propre protocole qui peut être différent de celui d'un autre cabinet médical.
À quelle fréquence doit-on surveiller les polypes de l'intestin grêle ?
Le type de test et la fréquence des tests de dépistage des polypes de l'intestin grêle dépendent du syndrome et des caractéristiques du patient. En général, le patient atteint du syndrome de Peutz-Jeghers doit faire l'objet d'une surveillance de l'intestin grêle (vérification périodique de la présence de polypes), car ces polypes peuvent provoquer une obstruction. Le gastro-entérologue de votre enfant conseillera une endoscopie par capsule ou une IRM spéciale de l'abdomen de l’enfant. La fréquence varie généralement entre 1 et 3 ans et est déterminée, en partie, par le nombre de polypes observés lors des interventions précédentes.
Quel type de chirurgie est recommandé pour les patients atteints de polypose – une colectomie partielle ou totale ?
Le patient atteint de polypose doit subir une colectomie pour prévenir le développement d'un cancer (dans le cas de la Polypose adénomateuse familiale) ou, dans certains cas, pour contrôler le nombre de polypes qui n'ont pas pu être enlevés par endoscopie (polypose juvénile ou syndrome de Peutz-Jeghers). Chez le patient atteint de la Polypose adénomateuse familiale, on prévoit soit une colectomie totale avec une poche, soit l'ablation du côlon moins le rectum auquel l'intestin grêle est rattaché – une anastomose iléoanale. Le choix du type de colectomie doit être fait en tenant compte de nombreux facteurs, notamment les antécédents familiaux, l'âge et le sexe de l'enfant et le type de mutation génétique, s'il est connu. Il est important de noter que, même après une colectomie, une surveillance continue de la partie restante de l'intestin est nécessaire pour détecter les polypes.
La calprotectine fécale a-t-elle un rôle à jouer dans la prédiction ou le suivi de la progression des polypes ?
La calprotectine fécale est une protéine qui peut être détectée lors d'un test de selles. Elle est lâchée par certaines cellules du système immunitaire dans l'intestin et peut être un marqueur d'inflammation de l'intestin. La calprotectine peut être utile pour prédire la présence de polypes, en particulier pour les polypes juvéniles, mais elle ne permet pas de faire la différence avec d'autres causes de douleurs abdominales ou de selles sanguinolentes, comme une infection ou une maladie inflammatoire de l'intestin. Des études complémentaires sont nécessaires pour déterminer si la calprotectine peut être utilisée pour prédire le développement ou la progression de polypes.
Les thérapies alternatives ou complémentaires jouent-elles un rôle dans le traitement des polypes ?
Il existe très peu d'études qui étudient les thérapies complémentaires dans le cadre de la polypose. Des études supplémentaires sont nécessaires pour déterminer si ces produits sont sûrs et efficaces chez les enfants.
Quel est le rôle du microbiome intestinal dans la formation et la progression des polypes ?
Il existe peu de preuves du rôle du microbiome intestinal dans la progression des polypes du côlon et le développement du cancer colorectal. Il n'existe à ce jour aucune indication diagnostique ou thérapeutique concernant le rôle du microbiome intestinal dans les troubles liés à la polypose.
La thérapie diététique joue-t-elle un rôle dans le traitement des polypes ?
De nombreuses études ont été menées sur des spécimens animaux atteints de la polypose adénomateuse familiale ainsi que sur des patients humains atteints d'adénomes à un âge plus avancé, mais aucune n'a été réalisée jusqu'à présent sur des enfants atteints de ce type de polypose. Une alimentation saine et équilibrée comprenant des fibres est importante, mais l'impact bénéfique de l'huile de poisson et des probiotiques, bien que probable, n'a pas encore été prouvé dans cette population. L'impact d'autres suppléments particuliers tels que le curcuma ou le resvératrol n'est pas non plus très clair et ils ne sont pas systématiquement recommandés.
Ressources
Comment trouver un gastroentérologue pédiatrique qui s'intéresse à la polypose ou qui est spécialisé dans ce domaine ?
Une équipe multidisciplinaire, composée d'un gastro-entérologue pédiatrique, d'un oncologue pédiatrique, d'un chirurgien pédiatrique et d'un conseiller en génétique, assure les meilleurs soins au patient atteint d’un syndrome de polypose.
Comment peut-on participer à la recherche ?
Demandez à votre gastroentérologue pédiatrique, oncologue et conseiller en génétique s'il existe davantage de possibilités de participer à la recherche dans votre centre.
Le vaccin COVID est-il sans risque pour l’enfant qui souffre de polypes ?
Oui, le vaccin COVID n'est pas une contre-indication et peut être injecté en toute sécurité.
Auteur : Thomas Attard, MD; Sabina Mir, MD; Isabel Rojas, MD; Mary Zachos, MD
Éditrice : Christine Waasdorp Hurtado, MD, MSCS, FAAP
Mars 2023
This post is also available in:
![]() English (Anglais)
English (Anglais) ![]() Español (Espagnol)
Español (Espagnol)

