
This post is also available in:
![]() English (Inglés)
English (Inglés) ![]() Français (Francés)
Français (Francés)
¿Qué es un pólipo?
- Un pólipo es un pequeño crecimiento de tejido extra que se forma a partir del revestimiento del intestino. A veces puede no haber síntomas cuando el pólipo es pequeño. Con el tiempo, los pólipos pueden crecer y ocasionar un bloqueo (obstrucción) en el intestino lo que puede causar dolor abdominal o vómito. En ocasiones, cuando se desprenden, pueden causar sangrado en las heces y un conteo bajo de glóbulos rojos (anemia).
- Los pólipos pueden formarse como resultado de mutaciones genéticas, que se trata de cambios en el ADN de la persona, los cuales pueden suceder al azar o por herencia y transmitirse de padres a hijos.
¿Cuándo debo llevar a mi hijo con su médico si estoy preocupado por un pólipo?
- Siempre se recomienda comentarle cualquier preocupación que tenga al proveedor de atención médica de su hijo. Por ejemplo, si su hijo experimenta dolor abdominal, vómito o sangre en las heces. También debe informar a su proveedor si existen antecedentes familiares de pólipos o cáncer de colon en un pariente cercano (ya sea en alguno de los padres, hermanos, abuelos, tías o tíos).
Poliposis Adenomatosa Familiar (PAF)
Diagnóstico
¿Qué es la PAF?
La poliposis adenomatosa familiar, conocida como “PAF”, es un síndrome de poliposis que causa cientos o miles de pólipos adenomatosos precancerosos en el tracto gastrointestinal, especialmente en el colon y recto. Con el tiempo, estos adenomas pueden transformarse en cáncer, por lo que se requiere seguimiento con endoscopía.
¿Qué causa la PAF?
En la mayoría de los casos, la PAF es causada por una mutación en el gen adenomatous polyposis coli (APC). Con frecuencia, esta mutación se hereda (se transmite en la familia). Sin embargo, 3 de cada 10 niños con PAF son los primeros en su familia en padecer PAF y, en algunos casos, no se identifica ninguna mutación. Los niños con PAF generalmente desarrollan múltiples pólipos adenomatosos cerca de la adolescencia – los pólipos adenomatosos son un tipo específico de pólipos que pueden volverse cancerosos con el tiempo. Los niños con pólipos adenomatosos se tratan de manera similar, independientemente si se identifica o no una mutación genética.
¿El fenotipo (presentación de la enfermedad) depende de la mutación genética?
Sí. Existen mutaciones en el gen APC que causan que se formen más pólipos colónicos con el tiempo. Las mutaciones generalmente se describen con un número que corresponde a la posición de la mutación (cambio en la secuencia de letras) dentro del gen. Estas incluyen mutaciones en ciertas áreas como el codón 1309 y entre los codones 1250 y 1464. También existe una forma más leve de PAF llamada PAF atenuada en la que los pólipos pueden formarse más adelante.
¿Existen otras afecciones asociadas con la PAF?
Además de los pólipos en el colon, las personas con PAF están propensas a desarrollar pólipos en otras partes del intestino. Los niños con PAF, corren el riesgo de desarrollar un tipo de tumor hepático (hepatoblastoma) fuera del intestino. También puede haber detecciones en la parte posterior del ojo - llamada hipertrofia congénita del epitelio pigmentario de la retina o lesiones pigmentadas del fondo ocular por PAF. Los niños también pueden tener crecimientos benignos en los huesos y la piel – osteomas y fibromas, y dientes extras o faltantes. Las personas con PAF también corren un mayor riesgo de padecer otros tipos de cáncer, tumores desmoides, cáncer de tiroides y tumores cerebrales.
¿Deben los familiares en primer grado de parentesco ser examinados para la PAF?
Sí. Se les debe ofrecer pruebas genéticas para la mutación APC identificada a todos los familiares en primer grado. Si no se han realizado pruebas genéticas o se desconoce la mutación genética, se recomienda una colonoscopía anual a partir de los 10 a 15 años de edad. Si no se encuentran adenomas después de los 15 años de edad, se puede realizar la colonoscopía cada 2 años o más.
Control
¿Cuándo debe realizarse la primera endoscopia de tracto gastrointestinal superior en un niño con PAF?
La endoscopía de tracto gastrointestinal superior se debe hacer alrededor de los 20 a 25 años de edad. Sin embargo, si existen antecedentes familiares de cáncer duodenal o síntomas preocupantes, se debe realizar una evaluación antes.
¿Cuándo debe someterse mi hijo a su primera colonoscopia?
Para los niños con PAF confirmada y para los niños en riesgo, en quienes no es posible realizar pruebas genéticas, la vigilancia del colon debe empezar entre los 12 y 14 años de edad. La PAF atenuada es una forma más leve de la enfermedad en la que los pacientes pueden no tener tantos adenomas y, por lo tanto, puede que no se presenten hasta la adultez.
¿Con qué frecuencia se debe realizar la vigilancia?
La colonoscopía de vigilancia debe realizarse cada 1 a 3 años dependiendo del fenotipo. La colonoscopía se recomienda antes, si el niño presenta sangre en las heces, anemia u otros síntomas de pólipos.
¿Cuál es el tratamiento para la PAF?
En la actualidad, no existe ningún tratamiento curativo para la PAF. Los niños con PAF necesitan una estrecha vigilancia de los pólipos que desarrollan cambios precancerosos para eliminarlos por medio de una endoscopia. Sin embargo, a veces hay demasiados pólipos que eliminar con una endoscopía y puede ser necesaria una cirugía.
¿Cuáles son los tipos de cirugías que se realizan para la PAF?
Existen 3 tipos de cirugías que se realizan en los niños con PAF, todas con la intención de extirpar la mayor parte del colon o el colon completo:
- Colectomía total con anastomosis ileorrectal (IRA, siglas en inglés)
- Proctocolectomía con anastomosis ileoanal con reservorio (IPAA, siglas en inglés))
- Proctocolectomía con ileostomía terminal
Debido a la infrecuencia del cáncer de colon en los niños, no se ha establecido el momento de la colectomía en niños y adolescentes menores de 18 años. Incluso después de la colectomía, la parte restante del recto o el reservorio (bolsa) debe ser monitoreado cada 6 a 12 meses para detectar el desarrollo de pólipos y cáncer.
¿Qué hay de otros tipos de cáncer?
Existe un mayor riesgo de cáncer de tiroides y se debe realizar una ecografía tiroidea durante la etapa final de la adolescencia y si resulta normal, repetirla cada 2 a 5 años. También existe una asociación con el hepatoblastoma, un tipo de cáncer de hígado. El examen abdominal, la ecografía y el control de los niveles de AFP en la sangre pueden realizarse cada 3 a 6 meses hasta los 5 años de edad.
No existe ninguna evidencia para llevar a cabo pruebas rutinarias en busca de tumores cerebrales o cáncer de páncreas. Dichas pruebas deben realizarse según los antecedentes familiares. Si bien, el cáncer de estómago e intestino delgado puede presentarse, no se recomienda una vigilancia especializada a menos que haya pólipos avanzados en el estómago o intestino delgado. Para los pacientes con PAF y antecedentes familiares de tumores desmoides, debe realizarse una resonancia magnética abdominal cada 3 a 5 años después de la colectomía para detectar tumores desmoides abdominales.
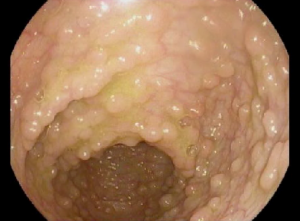
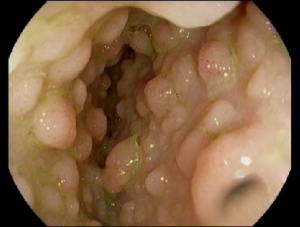
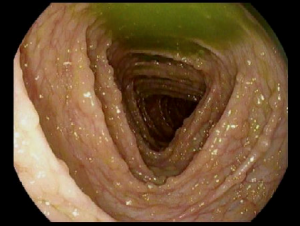
Síndrome de Peutz-Jeghers (SPJ)
Diagnóstico
¿Qué es el SPJ?
El síndrome de Peutz-Jeghers (SPJ) es una enfermedad hereditaria (transmitida de un miembro de la familia) poco común, que se presenta con pólipos, manchas de color oscuro, y un mayor riesgo de ciertos tipos de cáncer. Ocurre en aproximadamente 1 de cada 200,000 personas.
Los niños con SPJ pueden presentar pólipos en el tracto gastrointestinal (GI) (estómago, intestino delgado e intestino grueso) así como en la nariz, los pulmones y la vejiga. Estos pólipos se consideran pólipos hamartomatosos porque poseen características específicas cuando se observan bajo un microscopio.
Los pólipos hamartomatosos son sobrecrecimientos de tejido benignos (no cancerosos). El SPJ también se asocia con pecas de ciertas características o manchas oscuras (pigmentación mucocutánea) alrededor de los labios y otras partes del cuerpo (boca, fosas nasales, manos, pies, zona perianal) que pueden desaparecer con el tiempo.
El diagnóstico se hace al presentarse:
- 2 o más pólipos de SPJ en la endoscopía
- Cualquier número de pólipos de SPJ con antecedentes familiares del síndrome de Peutz-Jeghers
- Pecas con ciertas características (en los labios y lecho ungueal) y antecedentes familiares de SPJ
- Cualquier número de pólipos de SPJ y pecas peculiares
¿Qué causa el SPJ?
El SPJ es causado por una mutación (cambio) en el gen STK11 que es responsable de controlar el crecimiento celular. El SPJ se produce cuando existe una mutación en una copia de este gen; sin embargo, no todos los pacientes con SPJ tienen una mutación en el gen STK11. Aproximadamente, 1 de cada 2 niños con SPJ tiene un padre con el síndrome y la otra mitad de las veces los niños pueden ser los primeros afectados en su familia.
¿Depende el fenotipo (presentación de la enfermedad) de la mutación genética?
No existe una relación clara entre la mutación del gen (genotipo) y la presentación de la enfermedad (fenotipo). Sin embargo, existe una variabilidad entre los pacientes en cuanto al tiempo de la presentación de la enfermedad y el tipo de la enfermedad (fenotipo), la cual puede deberse al desarrollo de los pólipos. La presentación de la enfermedad puede incluir anemia, debido al sangrado gastrointestinal de los pólipos, dolor abdominal, obstrucción del intestino delgado (bloqueo) o intususcepción (donde el pólipo puede causar que el intestino se pliegue dentro de sí mismo) lo que provoca dolor, vómito y a veces, una obstrucción completa.
¿Existen otras afecciones asociadas al SPJ?
Los niños y los adultos con SPJ corren el riesgo de sufrir otras manifestaciones de la enfermedad fuera del intestino – en los niños, estas incluyen tumores testiculares y ováricos, para los cuales se necesitará un examen de ultrasonido periódicamente. En los adultos, también existe el riesgo de cáncer del cuello uterino, de mama, de ovario, de endometrio y de páncreas.
¿Deben realizarse pruebas de detección de SPJ en los familiares de primer grado?
Si. Se les debe ofrecer pruebas genéticas a todos los familiares de primer grado. En la gran mayoría de los pacientes con SPJ, se detecta la mutación del gen STK11, pero si no se ha detectado ningún gen en uno de los miembros de la familia, no se pueden realizar pruebas genéticas en los niños.
Control
¿Cuándo debe realizarse una endoscopia en un niño con SPJ?
Se recomienda que la revisión del tracto GI empiece a más tardar a los 8 años de edad, a menos que los síntomas aparezcan antes. Los estudios deberán incluir una endoscopía del tracto gastrointestinal superior (la cual permite examinar el esófago, el estómago, y la primera parte del intestino delgado) y una colonoscopía (la cual permite examinar el intestino grueso hasta la última parte del intestino delgado). Como no se puede observar todo el intestino delgado con el endoscopio, se utilizan otros métodos para hacerlo, como la resonancia magnética o la cápsula endoscópica (una pastilla con una cámara que se mueve a través del tracto GI).
¿Con qué frecuencia debe realizarse la vigilancia?
La vigilancia del tracto GI debe individualizarse en función a los síntomas, el riesgo de complicaciones de los pólipos versus el riesgo de las pruebas, y la perspectiva familiar. Es muy importante hablar con su consejero genético y su gastroenterólogo sobre los síntomas a los que debe prestar atención (sangrado, dolor abdominal y vómito) para que los niños sean evaluados cuanto antes si es necesario.
¿Cuál es el tratamiento para el SPJ?
Actualmente, no existe ningún tratamiento preventivo o curativo para el SPJ. Los niños con SPJ deben tener una vigilancia estrecha para detectar los pólipos que pueden causar complicaciones como sangrado, intususcepción u obstrucción. Si es posible, el objetivo es quitar los pólipos grandes para evitar estas complicaciones, lo cual se puede lograr con una endoscopía. Sin embargo, a veces las complicaciones pueden ocurrir inesperadamente y requerir cirugía inmediata.
Los pacientes con una obstrucción o intususcepción intestinal requieren cirugía de urgencia. Al momento de la cirugía es posible evaluar el resto del intestino delgado utilizando un endoscopio durante la cirugía para tratar de identificar y eliminar los pólipos grandes adicionales.
¿Qué pasa con el riesgo de cáncer asociado al SPJ?
El cáncer durante la infancia relacionado al SPJ es poco frecuente. Desafortunadamente, existe un mayor riesgo de cáncer de por vida a partir de la edad adulta. En los niños, es importante realizar controles y exámenes físicos regulares para detectar signos de pubertad precoz que puedan indicar un tumor ovárico poco común (niñas) o testículos (niños). Para los niños, el examen testicular anual y la observación de cualquier signo de pubertad precoz o aumento del tamaño de las mamas deben comenzar al momento del diagnóstico.
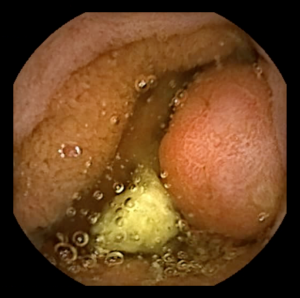
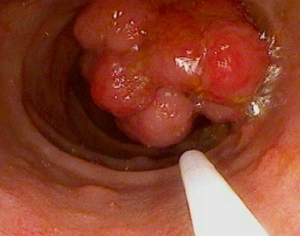
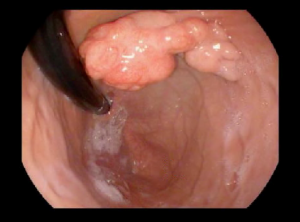
Síndrome de Poliposis Juvenil
Diagnóstico
¿Qué es el síndrome de poliposis juvenil?
El síndrome de poliposis juvenil es una afección poco común que resulta en la formación de pólipos juveniles (hamartomatosos) en el tracto gastrointestinal (GI) y se asocia con un mayor riesgo de cáncer colorrectal. Afecta aproximadamente a 1 de cada 130,000 personas.
Este síndrome difiere de los pólipos juveniles solitarios, que son el tipo más común de pólipos en los niños y no causan un riesgo de cáncer asociado.
El diagnóstico de la poliposis juvenil, se lleva a cabo cuando se encuentran 5 o más pólipos juveniles en el colon o recto, o en una persona con antecedentes familiares de síndrome y cualquier número de pólipos juveniles o cualquier pólipo juvenil que se encuentre en otras partes del tracto GI.
¿Qué causa la poliposis juvenil?
La poliposis juvenil causada por un defecto (una mutación) en la función de un gen. Existen dos mutaciones implicadas, SMAD4 en el cromosoma 18 y BMPR1A en el cromosoma 10. Se hereda en un patrón autosómico dominante, lo que significa que se transmite de generación en generación. Sin embargo, solo aproximadamente 1 de cada 2 niños presenta un defecto en sus genes. El otro 50% cumple los criterios para el diagnóstico, pero no se identifica una mutación genética en las pruebas.
Los pacientes sin una mutación genética identificada tienen pocos o ningún pólipo en el estómago y el intestino delgado, y el número de pólipos en el colon disminuye después de los primeros 10 años de edad. Estos pacientes también suelen presentar la enfermedad a una edad más temprana, tienen antecedentes familiares negativos y un menor riesgo de resección del colon.
¿Depende el fenotipo (la presentación de la enfermedad) de la mutación genética?
Si. Aunque todavía no está claro, los pacientes con la mutación SMAD4 tienen más pólipos en el estómago y un mayor riesgo de presentar cáncer. Además, algunos pacientes con esta mutación genética pueden tener lo que se llama telangiectasia hemorrágica hereditaria (HHT, siglas en inglés). Estos pacientes tienen vasos sanguíneos anormales en varios órganos incluyendo el cerebro, los pulmones y el hígado y usualmente presentan sangrados nasales, difíciles de controlar.
Existe una forma grave de poliposis, conocida como poliposis juvenil de la infancia (PJI). Estos pacientes también presentan otra mutación en el mismo cromosoma 10 llamada PTEN. Se presenta durante los primeros 2 años de vida y es más agresiva cuando aparece: con diarrea, proteínas bajas, sangrado intestinal, poca ganancia de peso y generalmente requiere la resección del colon a una edad más temprana.
¿Existen otras afecciones asociadas?
Actualmente no se conocen afecciones alérgicas o autoinmunes asociadas con la poliposis juvenil.
¿Deben ser evaluados los familiares de primer grado para el síndrome de poliposis juvenil?
Si. Cuando se ha identificado una mutación genética, los miembros de la familia se deben hacer pruebas. Si no se identifica ninguna mutación, se recomienda que los padres y hermanos mayores de 15 años de edad, se realicen una colonoscopía para evaluar la presencia de pólipos juveniles.
Control
¿Cuándo se debe realizar una endoscopia en un niño con poliposis juvenil?
Se recomienda empezar la endoscopía de tracto gastrointestinal inferior (colonoscopia) a partir de los 12-15 años de edad. La endoscopía superior no es necesaria antes de los 18 años de edad, a menos que los síntomas aparezcan antes.
¿Con qué frecuencia se debe realizar la vigilancia?
La vigilancia es necesaria para prevenir sangrado, anemia, dolor abdominal y para la prevención de cáncer. Se recomienda realizar una colonoscopía anual si se encuentran pólipos y hasta que se hayan eliminado todos los pólipos grandes, y después cada 1 a 5 años.
¿Cuál es el tratamiento para la poliposis juvenil?
No existe ningún otro tratamiento específico para este síndrome, aparte de la resección de los pólipos, también llamada polipectomía.
En los pacientes con la forma grave (SPI), se ha demostrado que un medicamento llamado sirolimus, mejora la anemia y los niveles bajos de albúmina y reduce el riesgo de la extirpación del colon.
¿Qué pasa con el riesgo de cáncer asociado con la poliposis juvenil?
En general, el riesgo de transformación maligna en los niños con poliposis juvenil es poco común, pero el riesgo de presentar cáncer de colon o recto aumenta un 10 a 50% de por vida. Por lo tanto, es importante la vigilancia con colonoscopía y la resección de los pólipos grandes. Los niños con la forma grave (SPI) corren un mayor riesgo de presentar cáncer de tiroides, cáncer de mama, cáncer de endometrio y otros tipos de cáncer en un futuro debido a la mutación del gen PTEN y se recomienda iniciar vigilancia con ecografía de tiroides a los 7 años de edad.
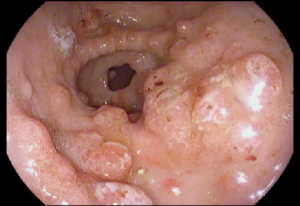
Preguntas generales y diversas
¿Existe un régimen estándar para la limpieza intestinal antes de una colonoscopia?
Se requiere una preparación intestinal antes de una colonoscopía. No hay un protocolo estándar universal en pediatría. Cada programa de gastroenterología pediátrica crea su propio protocolo el cual podría diferir del de otros programas.
¿Con qué frecuencia debe realizarse la vigilancia de los pólipos del intestino delgado?
El tipo de prueba y la frecuencia de su realización para los pólipos del intestino delgado dependen del síndrome y las características relacionadas con el paciente. En general, los pacientes con síndrome de Peutz-Jeghers necesitan vigilancia del intestino delgado (chequeos periódicos para detectar pólipos) porque estos pólipos pueden causar una obstrucción. Su gastroenterólogo lo orientará para realizar una endoscopía capsular en su hijo o una resonancia magnética especial del abdomen. La frecuencia varía entre 1 y 3 años normalmente y se determina, en parte, por la cantidad de los pólipos que se observaron en procedimientos anteriores.
¿Qué tipo de cirugía, colectomía parcial o total, se recomienda para los pacientes con poliposis?
Los pacientes con poliposis requieren una colectomía para prevenir el desarrollo de cáncer (en PAF) o en algunos casos para controlar el número de pólipos que no se pudieron eliminar con endoscopía (poliposis juvenil o síndrome de Peutz-Jeghers). Para los pacientes con PAF, se planifica una colectomía total con reservorio (IPAA) o una resección del colon, sin incluir el recto, al cual se le conecta el intestino delgado- anastomosis ileoanal (IRA). La decisión sobre el tipo de colectomía, se debe tomar teniendo en cuenta varios factores incluyendo los antecedentes familiares, la edad y el género del niño y el tipo de mutación genética, si se conoce. Es importante tener en cuenta que, incluso después de la colectomía, es necesaria una vigilancia continua de la parte restante del intestino para detectar pólipos.
¿Juega algún papel la calprotectina fecal en la predicción o el seguimiento de la progresión de pólipos?
La calprotectina fecal es una proteína que se puede detectar en un análisis de heces, es liberada por algunas células que se encuentran en el sistema inmunológico del intestino y puede ser un marcador de inflamación en los intestinos. La calprotectina puede ser útil para predecir la presencia de pólipos, en especial los pólipos juveniles. Sin embargo, no ayuda a distinguir las causas del dolor abdominal o heces con sangre por infección o la enfermedad inflamatoria intestinal. Se necesitan más estudios para determinar si se puede utilizar la calprotectina para predecir el desarrollo o la progresión de pólipos.
¿Qué tal son las terapias alternativas o complementarias para el tratamiento de los pólipos?
Muy pocos estudios de investigación han evaluado las terapias complementarias en la poliposis. Se necesitan más estudios para determinar si son seguras y eficaces para ser utilizadas en los niños.
¿Qué papel juega el microbioma intestinal en la formación y progresión de los pólipos?
Existen pruebas limitadas referentes al papel que juega el microbioma intestinal en la progresión de los pólipos colónicos y el desarrollo de cáncer colorrectal. En la actualidad, no existen implicaciones diagnósticas o terapéuticas del microbioma intestinal en los trastornos de poliposis.
¿Qué tal es la terapia dietética para los pólipos?
Se han llevado a cabo varios estudios en modelos animales de PAF, así como en pacientes con adenomas a una edad más avanzada, pero hasta ahora, ninguno en niños con PAF. Una dieta saludable y balanceada que incluya fibra es importante, pero el efecto beneficioso del aceite de pescado y los probióticos, aunque probable, sigue sin demostrarse en esta población. El efecto de otros suplementos específicos como la cúrcuma o el resveratrol tampoco está claro y no se recomienda de forma rutinaria.
Recursos
¿Cómo encuentro un gastroenterólogo pediatra con interés o experiencia en poliposis?
El cuidado óptimo para los pacientes con el síndrome de poliposis incluye un equipo multidisciplinario compuesto por un gastroenterólogo pediatra, un oncólogo pediatra, un cirujano pediatra, y un consejero genético.
¿Cuáles son las oportunidades para participar en estudios de investigación?
Consulte a su gastroenterólogo pediatra, oncólogo y consejero genético acerca de las oportunidades adicionales para participar en estudios de investigación en su centro.
¿Es segura la vacuna contra el COVID para los niños con pólipos?
Si. La vacuna contra el COVID no es perjudicial y puede administrarse de manera segura.
Autor: Thomas Attard, MD; Sabina Mir, MD; Isabel Rojas, MD; Mary Zachos, MD
Editor: Christine Waasdorp Hurtado, MD, MSCS, FAAP
Marzo 2023
This post is also available in:
![]() English (Inglés)
English (Inglés) ![]() Français (Francés)
Français (Francés)

