This post is also available in:
![]() Español (Spanish)
Español (Spanish) ![]() Français (French)
Français (French)
What is Wilson’s Disease?
Wilson’s disease is a rare, inherited disorder caused by the inability of the body to properly excrete copper. The copper abnormally builds up in the organs (most commonly in the liver, brain, and eyes) and causes symptoms specific to which organs are damaged. If not treated effectively, there can be life-threatening complications.
How common is Wilson’s Disease?
Wilson’s disease is rare and affects 1 out of 30,000 individuals. It is more common in people who have a family history of the condition. Most people are diagnosed between the ages of 5-35 years.
What are the symptoms of Wilson’s Disease?
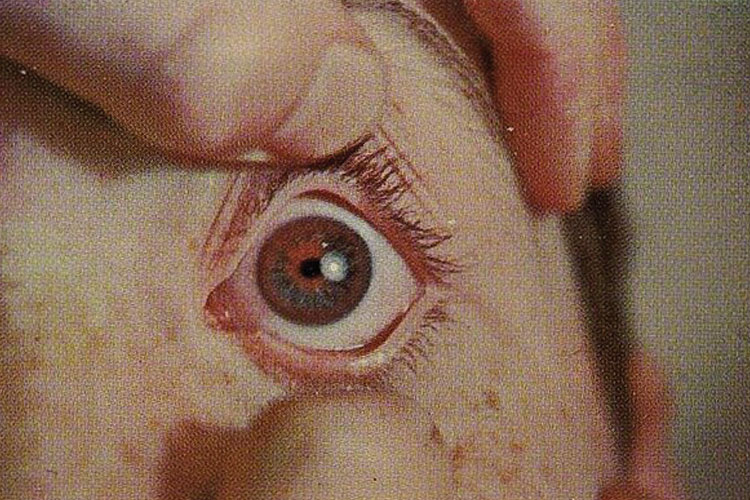
Symptoms appear once there is a high amount of copper in the organs that are affected. If the liver has been affected, people can have yellowing of the skin (jaundice) or the eyes (icterus), loss of appetite, fatigue, dark urine or light stool, or abdominal swelling or bloating from fluid buildup. Brain involvement can cause difficulty with speech, swallowing, or physical coordination, and changes such as muscle stiffness or uncontrolled movements. People can also experience depression or other mood and personality changes. Copper-colored rings may also be present in the front of the eye (also known as Kayser-Fleischer rings); these are usually only visible with an eye exam. The heart, kidneys, blood, and joints may also be involved. Finally, some patients with Wilson’s Disease have no symptoms and are diagnosed because there is a family history of the disease.
What causes Wilson’s Disease?
Wilson’s disease is inherited and passed down to the affected person through the family. It is caused by changes to the ATP7B gene; this change prevents the body from getting rid of extra copper in the body. Instead of sending copper from the liver to other parts of the body through bile, the liver stores the copper, where it eventually builds up and causes damage to the liver. While copper starts to build up at birth, symptoms do not appear until later in life.
How is Wilson’s Disease diagnosed?
Wilson’s Disease can be difficult to diagnose because of the number of bodily organs affected, the resulting variety in symptoms, and its resemblance to other diseases. Most commonly, Wilson’s is diagnosed with a combination of a medical and family history, a physical exam including an eye exam, and blood and urine tests. Imaging of the liver with an MRI or ultrasound may also be recommended. During these procedures, a biopsy of the liver will be performed by removing a small piece of liver tissue with a needle. Genetic testing can also confirm Wilson’s Disease.
How is Wilson’s Disease treated?
Wilson’s Disease requires lifelong treatment to manage symptoms and prevent further organ damage. When diagnosed and treated early, most people with the condition live normal lives. It can be treated with zinc, which blocks the absorption of more copper from the intestines. There are also medicines that help the body eliminate the excess copper, known as chelating agents. Diet adjustments are also recommended to avoid high copper-containing foods (including chocolate, liver, mushroom, nuts, and shellfish). In certain, more severe cases (such as liver failure), liver transplant surgery may be required.
Authors: Vania Kasper, MD and Shivany Pathania, MD
Editor: Amanda Deacy, PhD
January 2024
This post is also available in:
![]() Español (Spanish)
Español (Spanish) ![]() Français (French)
Français (French)

